
ImprintPocket: a program of generation of binding site negative images
Zheng Ouyang
The main aim of this study is to develop and
implement ˇ°ImprintPocketˇ±, an algorithm for the rapid, accurate
and automated identification negative image (pseudoligand) of binding pocket in
protein structures for structure-based virtual screening. The program to perform a negative image search is an
extension of CASTp, a suite of programs for locating pockets and cavities in protein crystal structures and
quantifying their size, which developed by Jie Liang et al. . The method is a computational geometry treatment of complex
shapes, based on alpha shape and discrete flow theory. ImprintPocket primarily developed
for proteins, but the algorithm is sufficiently robust to allow the analysis of
any molecular system, including nucleic acids or inorganic material.
Calculations can be performed using discrete structures from crystallographic
analysis and NMR experiments as well as with trajectories from molecular
dynamics simulations. It has be integrated into a new structure-based
pharmacophore search method (called Shape4). ImprintPocket is freely available from the web site http://sts.bioengr.uic.edu/pni/.
Based on alpha-shape and discrete-flow methods [1-3], ImprintPocket creates pocket negative images follow seven steps, (1) identifies the atoms forming pockets, (2) computes volume and area of pockets, (3) identifies atoms forming ˇ°rimsˇ± of the pocket mouth(s), (4) computes the number of mouth openings for each pocket, (5) computes the area and circumference of mouth openings, and (6) locates cavities and measures their size. (7) builds pocket negative image from pocket tetrahedrons. The pocket prediction process specifies the calculation of the socalled "dual complex" (or alpha shape) and is summed up for a simplified two-dimensional depiction of binding site atoms. The procedure includes the calculation of the Voronoi diagram which consists of Voronoi cells. Each Voronoi cell contains one protein atom and controls all spatial points that are closest to the respectively considered atom. The Voronoi diagram is mathematically equivalent to the Delaunay triangulation of the complex hull drawn around the protein atom centers. To obtain the dual complex, Voronoi edges and vertices are disregarded in the triangulation, if they are situated completely or in part outside of the molecule. A triangle with one or more omitted edges is denoted as "empty". Neighboring empty triangles are combined in the "disrete-flow" method to outline continuous voids in the protein surface. In the course of this process an obtuse empty triangle flows to its neighboring triangle, whereas acute empty triangles act as sinks to collect the flow of neighboring triangles
Methods for
extracting binding site negative image have been developed and used in various
docking programs. For example, DOCK16 uses a process that places spheres into the
binding site randomly followed by clustering analysis to identify the best set
of spheres to represent the shape of the binding site. Surflex24 uses a
different approach to generate so-called ˇ°prototype moleculesˇ± to represent the
binding site shape and other information. Our approach is unique in that it
uses alpha-shape to deterministically detect the binding site atoms, followed
by a geometric casting algorithm to generate the negative image as a collection
of spheres. Here, we define a pocket negative image as a set of circumscribed
spheres derived from the discrete set of Delaunay tetrahedra and triangles for
a pocket. For a tetrahedron abcd,
there is a unique point z, the
orthogonal center, that has the same power distance to the four atom centers at
points a, b, c, and d.
Figure 1: 2D representation of
orthogonal center. C is orthogonal
center;
r is power
distance; V0 , V1 , V2 are atom 1,2,3
respectively.
The center tangent spheres are used as circumscribed spheres for pocket triangles.
Overlap checking is performed to prevent circumscribed spheres from overlapping
with other pocket atoms. We also set a threshold to remove tiny spheres. The remaining
circumscribed spheres thus make up the negative image of the ligand pocket. According
the pocket size, certain number of top largest negative images will be output
in PDB format. Each negative image is composed of points and their radius.
The method was implemented in C++ on both Linux and Windows.
The program uses the publicly-licensed software newmat for matrix calculation. The core functions of CASTp
are integrated in this program. Based on the testing results of 50 protein
structures, the calculation time for most structure is less than 2 minutes. A
web server (http://sts.bioengr.uic.edu/pni/) was setup for users upload target structure for negative
image computing, and the result files will be packed and emailed back or use
can download it online. It allows multiple structures uploading as long as user
compresses them into one file. A stand alone negative image viewer is available
for Windows uses. With a user-friendly GUI, user can visualize the output
easily. The program performance was tested on Pentium IV 1.6 GHz machine with 1
GB RAM running on Windows XP Professional operating system and AMD
Sempron(tm) machine with RedHat 8.0.
A new algorithm
for the identification of negative images of surface pocket in large molecular
systems was developed and implemented in the ImprintPocket program, which is available within the public domain. Under
this method, the negative image of the ligand-binding pocket of the target
protein is generated using virtual atoms, and with the expectation that it
could represent an optimal ligand of the target protein. The algorithm
automatically explores all kinds of surface cavities. Rigorous shape
representation and adjustable details of level provide user an accurate and
customizable tool for various structure-based researches. The user needs only
to provide the molecular PDB file and probe radii to enable the analysis of any
molecular system. The algorithm is sufficiently rapid and robust for the
routine analysis of large number of structures analysis. Here is an example of p38 MAP kinase and RO3201195, an orally bioavailable and
highly selective inhibitor of p38 which was selected for advancement into Phase
I clinical trials. The binding pocket and the negative image are illustrated in
Figure
39,
Figure
40.
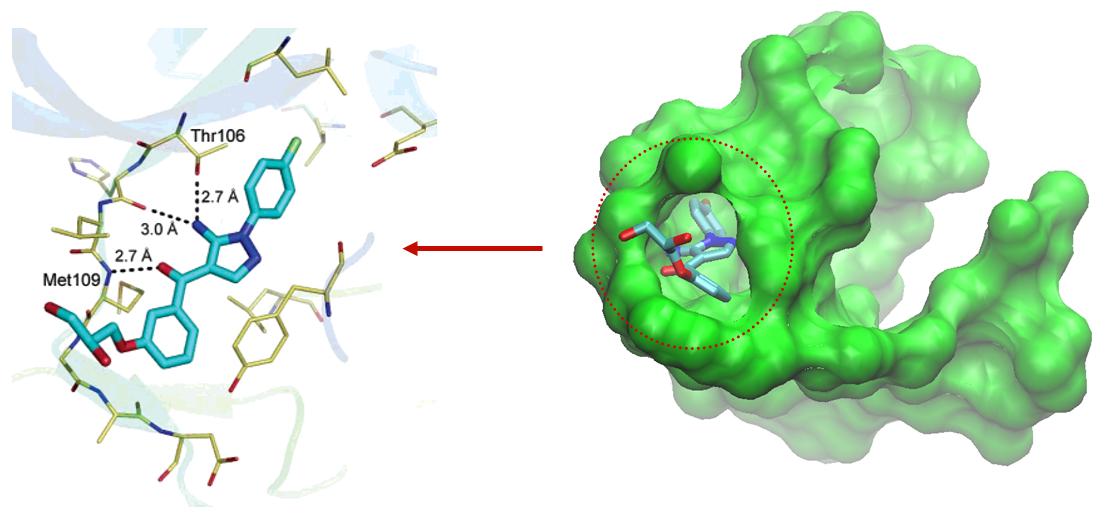
Figure 2: Mitogen-activated
protein kinase (2gfs) and inhibitor
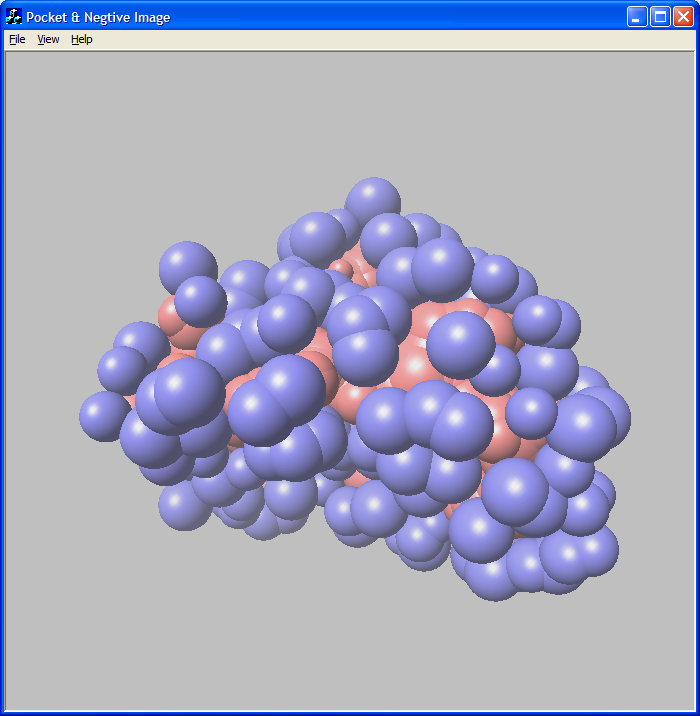
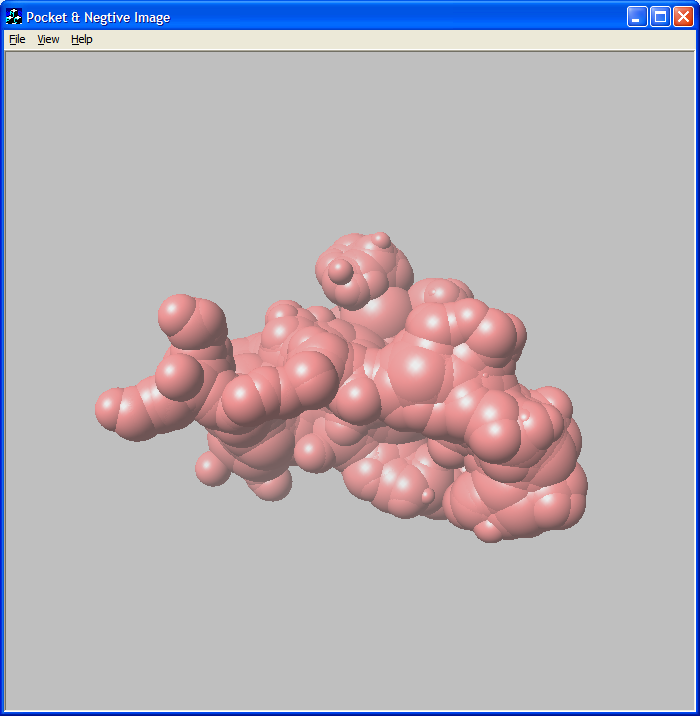
Figure 3: Mitogen-activated protein kinase (2gfs)
pocket and negative image.
Blue spheres are pocket atoms and red spheres are negative image atoms.
ImprintPocket has been
used to develop a new virtual screening tool - Shape4. Negative image is further
represented as Gaussian functions using the Shape toolkit (from OE Scientific)
and fast shape overlays between the negative image and database molecules can
be performed. Such an implementation can capture the intricate details of a
binding site shape, the effect of which during virtual screening experiments
has been demonstrated by five test cases in this work. Shape4 performed very
well in all virtual screening experiments and in some cases significantly
better than other virtual screening methods (ROCS and FRED) studied in this
work. Shape4 offers a fast, effective and intuitive virtual screening
alternative in cases where the X-ray crystal structure of the target is known
without performing computationally more expensive docking calculations.
1. Tropsha, A. and H. Edelsbrunner, Biogeometry: applications of computational geometry to molecular structure. Pac Symp Biocomput, 2005: p. 1-3.
2. Natarajan, V. and H. Edelsbrunner, Simplification of three-dimensional density maps. IEEE Trans Vis Comput Graph, 2004. 10(5): p. 587-597.
3. Edelsbrunner, H. and P. Koehl, The weighted-volume derivative of a space-filling diagram. Proc Natl Acad Sci U S A, 2003. 100(5): p. 2203-2208.